
Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) - Alles Wissenswerte zur Verordnung über In-vitro-Diagnostika
Inhalte, Timelines, Änderungen oder Übergangsfristen kompakt zusammengefasst
Die Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR)vom 5. April 2017 ist am 26. Mai 2022 in Kraft getreten. Die bis dahin gültige EU-Richtlinie für IVD, die Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD), wurde damit abgelöst und die IVDR bringt tiefgreifende Änderungen mit sich. Das heißt jedoch nicht, dass der regulatorische Schalter für Hersteller von IVD am 26. Mai 2022 vollständig umgelegt wurde. Die Änderungen sind komplex und auch bereits CE-gekennzeichnete IVD sind nach dem Anforderungen der IVDR neu zu zertifizieren. Die neue EU-Verordnung führt für viele Produkte zu höheren Anforderungen.Um das System nicht zum Kollabieren zu bringen, geschieht die Umsetzung der EU-Verordnung schrittweise unter Einbindung von Übergansfristen. Die wesentlichen Änderungen zusammengefasst:
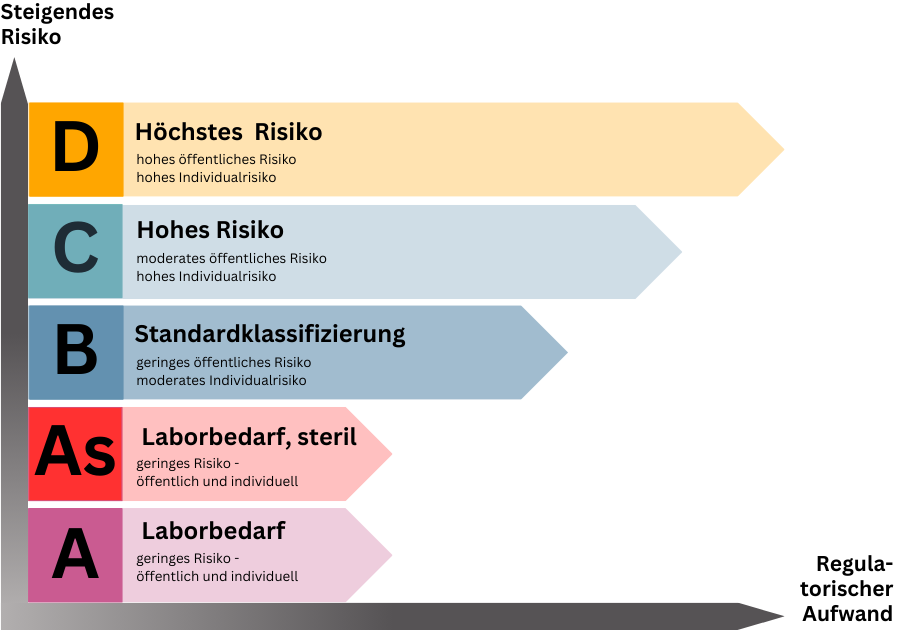
Einteilung in Risikoklassen
Die Klassifizierung sieht eine regelbasierte Einteilung in Risikoklassen von A/A steril (As) bis D bzw. geringem bis hohem Risiko vor. Eine vorab festgelegte Liste nach Klassen und Produkten gibt es leider nicht, sodass die Klassifizierung durch den Hersteller auf Basis des Regelwerkes zu erfolgen hat. Vom Hersteller falsch klassifizierte Produkte führen zu Abweichungen im Audit oder bei der Produktprüfung durch die Benannte Stelle, was den Zulassungsprozess und die Markteinführung empfindlich verzögern könnte. Weitere Informationen zur Einteilung der Risikoklassen und den 7 Klassifizierungsregeln gibt es hier.
Ohne Benannte Stelle geht (fast) nichts mehr
Über kurz oder lang ist nach Ablauf der Übergangsfristen für alle Produkte der Klassen A steril (As), B, C und D das Mitwirken einer Benannten Stelle für den Zulassungsprozess nach IVDR erforderlich. Lediglich nicht sterile Klasse A-Produkte sind davon ausgenommen. Die grundsätzliche Einbindung Benannter Stellen unter IVDR führt zu einem ungleich höheren Bedarf an Benannten Stellen als noch unter IVDD. Darüber hinaus wächst der Anspruch an die Benannten Stellen selbst, sodass Angebot und Nachfrage in ein erhebliches Ungleichgewicht rutschen könnten. Übersicht der Bennannten Stellen nach IVDR
Übergangsfristen der IVDR – keine Regel ohne Ausnahme
Die Übergangfristen hängen von der Risikoklasse ab. Welche Übergangsfristen für welche Risikoklassen zum Tragen kommen und welche Sonderfälle es gibt, erklären wir hier. Im Prinzip ist zu sagen, dass steigendes Risiko zu sinkenden Fristen führt - außer für nicht sterile Klasse A-Produkt. Für diese Produktklasse wird gemäß IVDR nach Konformitätsbewertung ohne Benannte Stelle eine Konformitätserklärung durch den Hersteller abgegeben, die in Folge zur CE-Kennzeichnung führt. Seit Inkrafttreten der IVDR am 26. Mai 2022 wird bereits so verfahren. Für die weiteren Risikoklassen gelten folgende Übergangsfristen:
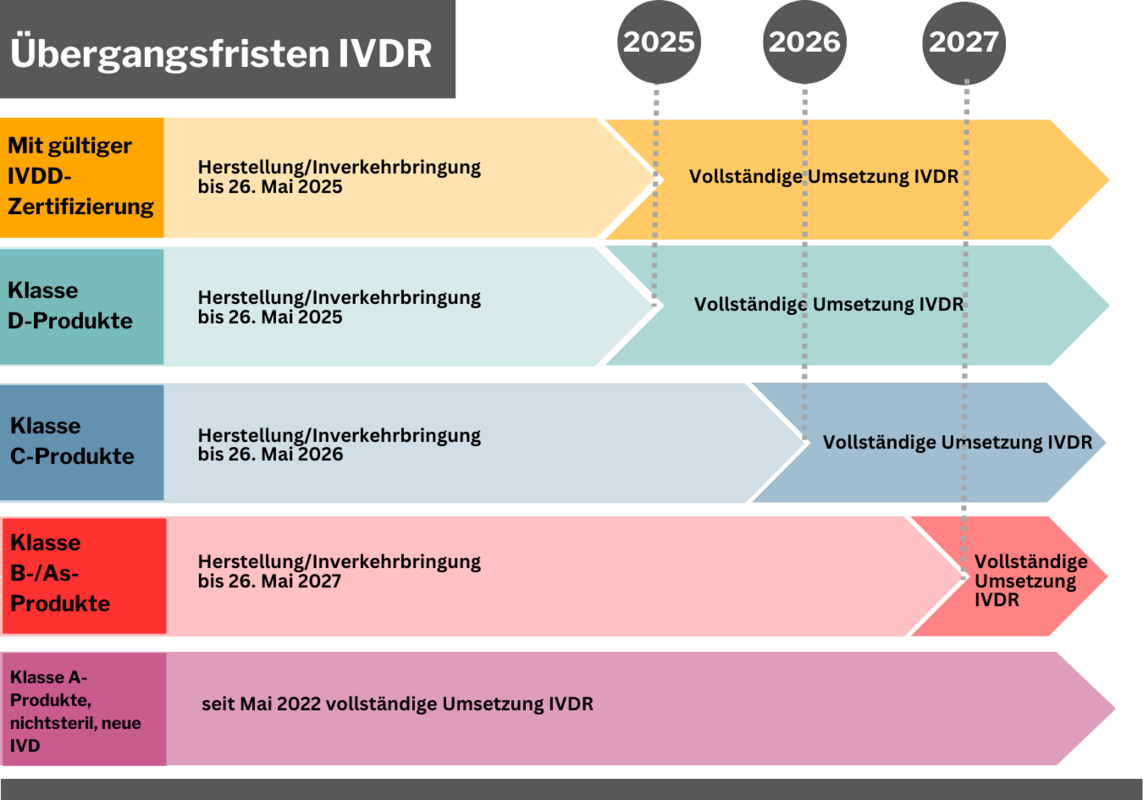
Produkte mit gültiger IVDD CE-Kennzeichnung, für die keine CE-Kennzeichnung nach IVDR angestrebt wird, dürfen weiter Inverkehr gebracht werden, sofern eine Benannte Stelle bei der IVDD CE-Kennzeichnung involviert war. Mit Ablauf der IVDD-Bescheiningung endet diese Möglichkeit jedoch.
Anforderungen bezüglich der Überwachung nach dem Inverkehrbringen (Post-Market-Surveillance), der Marktüberwachung, die Vigilanz und die Registrierung von Wirtschaftsakteuren der Produkte müssen umgehend gemäß IVDR umgesetzt werden und die Anpassung des Qualitätsmanagementsystems darf nicht außer Acht gelassen werden.
Verantwortliche Person zwingendes Muss – Person Responsible for Regulatory Compliance (PRRC)
Der Artikel 15 der IVDR sieht den Einsatz einer solchen verantwortlichen Person vor, daher wird in dem Zusammenhang auch häufig von einer Artikel-15-Person gesprochen. Im Konkreten heißt das, dass die Hersteller mindestens eine Person im Unternehmen benennen müssen, die für Einhaltung und Umsetzung der Regulierungsvorschriften verantwortlich ist. Der nahliegende Gedanke, dass die PRRC der neue Name für die Sicherheitsbeauftragten ist, ist nicht zutreffend. Verantwortung und Befugnisse gehen weit über die bisherigen hinaus. Die Qualifikation der Person Responsible for Regulatory Compliance (PRRC) ist zwingend nachzuweisen und darüber hinaus ist die Registrierung in der EUDAMED und im Actor Registration Module verpflichtend. Zusätzlich ist eine freiwillige Registrierung im Deutsches Medizinprodukte-Informations- und Datenbanksystem (DMIDS) möglich. Neben Artikel 15 bietet die MDCG 2019-7_Rev.1 detailliertere Informationen bezüglich der erforderlichen Qualifikationen sowie der Aufgaben des PRRC.
Unique Device Identification (UDI)
Die schon aus MDR bekannte UDI, die einmalige Produktkennung bestehend aus einem numerischen oder alphanumerischen Code, die sich aus jeweils einmaligen Produkt- und Herstellungskennungen (Device Identifier = UDI-DI, Production Identifier = UDI-PI) zusammensetzt, ist nach IVDR für alle IVD verpflichtend. Die UDI ermöglicht effizientere Rückverfolgbarkeit sowie vereinfachte Rückrufe der Produkte. Die Fälschung von Medizinprodukten wird erschwert und die Patientensicherheit erhöht. Weitere Informationen zur UDI
Technische Dokumentation praktisch nicht ohne Leistungsstudie möglich
Die Leistungsanforderungen an die Technische Dokumentation ist erheblich gestiegen sowohl im Inhalt als auch im Umfang. Leistungsstudien sind gemäß IVDR erforderlich zur Feststellung oder Bestätigung der Analyseleistung und der klinischen Leistung eines Produktes. Die Hersteller haben im Rahmen der Technischen Dokumentation Nachweise zu bringen, dass die Risikoklasse der Art und der Sicherheit des Produktes entspricht.
Konformitätsbewertungsverfahren mit Benannter Stelle
Die Konformitätsbewertungsverfahren sind von der Risikoklasse abhängig und bei einer Benannten Stelle zu beantragen. Nichtsterile Klasse A-Produkte sind davon befreit. Es reicht eine Konformitätserklärung ohne Einbindung einer Benannten Stelle. Für alle weiteren Risikoklassen regeln die Anhänge I, II, III, IX, X und XI der IVDR die Konformitätsbewertungsverfahren. Auch hier steigt der Anspruch mit dem Risiko.
Risikoklasse As, B, C, D
- Anhang I – Grundlegende Sicherheits- und Leistungsanforderungen
- Anhang II – Technische Dokumentation
- Anhang III – Technische Dokumentation über die Überwachung nach dem Inverkehrbringen/Postmarket Suveillance
- Anhang IX – Qualitätsmanagementsystem
Risikoklasse C, D
- Anhang X – EU-Baumusterprüfung
- Anhang XI – Produktionsqualitätssicherung
- Anhang XI – Produktchargenprüfung
Weitere Infos zum Konformitätsbewertungsverfahren, wie sich die Anhänge auf die Risikoklassen auswirken
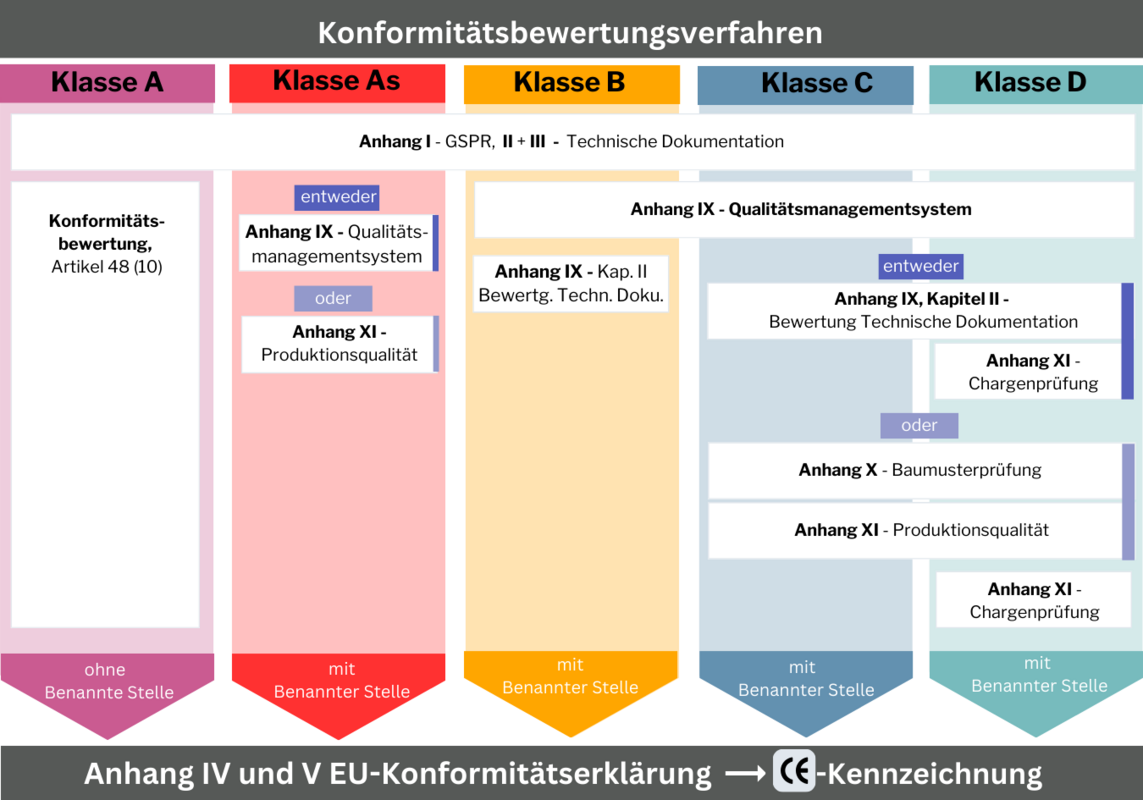
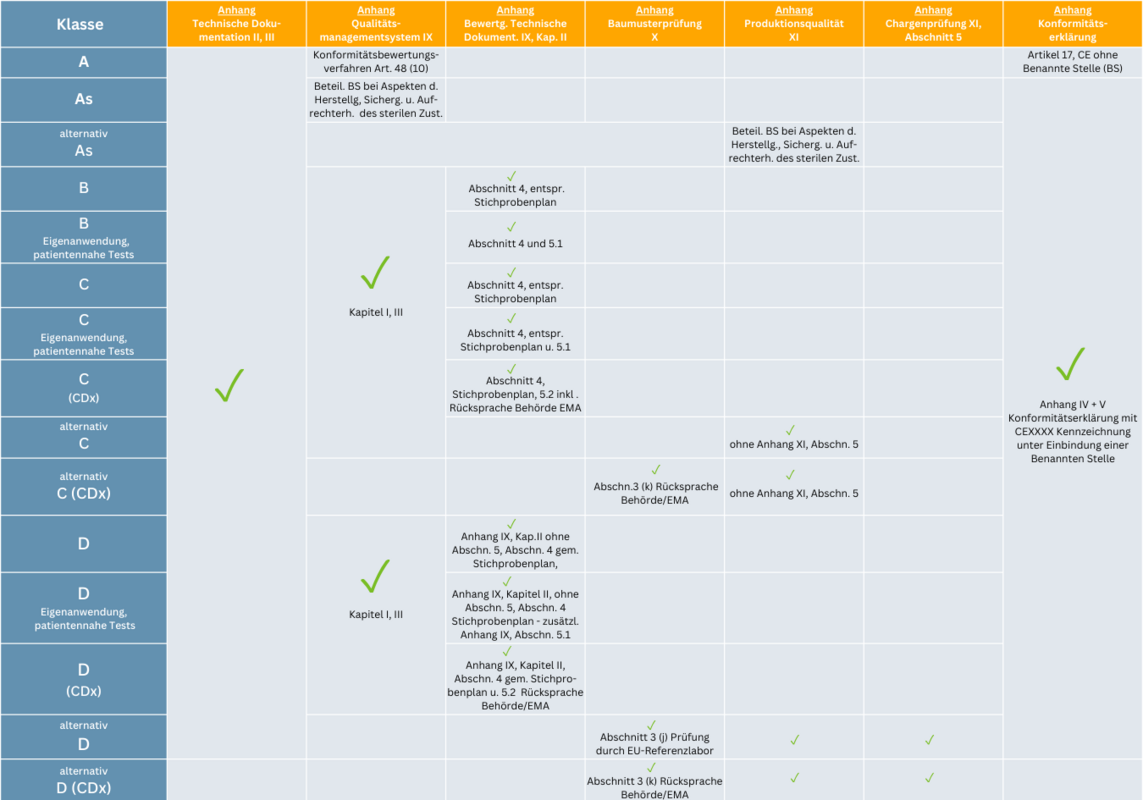
Risikoklassen und Anhänge im Überblick
Welche Anhänge für welche Risikoklasse von Bedeutung sind und im Rahmen des Konformitätsbewertungsverfahrens erfüllt sein müssen:
Nachweise zu den Grundlegende Sicherheits- und Leistungsanforderungen/ General Safety And Performance Requirements (GSPR)
Der Anhang I der IVDR regelt den Umgang mit den GSPR. Im Rahmen des Konformitätsbewertungsverfahren haben die Hersteller den Nachweis der Erfüllung der Grundlegenden Sicherheits- und Leistungsanforderungen zu erbringen u.a.:
- Risikomanagement, das ein vertretbares Nutzen-Risikoverhältnis gewährleistet
- Einhaltung der relevanten Normen (State of the Art)
- Leistungsbewertung
- Funktionale Sicherheit
- Biologische Sicherheit
- IT-Sicherheit (Cybersecurity)
- Software-Lebenszyklus-Prozesse einschließlich Verifizierung und Validierung
- Gebrauchsanweisungen etc.
Die dafür relevante Prüfnorm ist die IEC 61010-1 inklusive des dazugehörigem Particular Standard 61010-2-101. Der Nachweis dieser grundlegenden Anforderungen der IVDR durch ein akkreditiertes Prüflabor führt zwangsläufig zu einem widerspruchsfreien Ergebnis und damit zur Akzeptanz bei Benannten Stellen. Das KEYMKR Prüflabor ist für die im Rahmen der IVDR relevanten Sicherheitsprüfungen akkreditiert.
Technische Dokumentation und akkreditierte Prüfung - bei KEYMKR aus einer Hand.
IEC 61010-1
Sicherheitsbestimmungen für elektrische Mess-, Steuer-, Regel- und Laborgeräte - Teil 1: Allgemeine Anforderungen
IEC 61010-2-101
Sicherheitsbestimmungen für elektrische Mess-, Steuer-, Regel- und Laborgeräte- Teil 2-101: Besondere Anforderungen an In-Vitro-Diagnostik Medizingeräte
